En la actualidad y sobre todo tras la entrada en vigor del Anexo 1 sobre Fabricación de Medicamentos Estériles de las Normas de Correcta Fabricación de la Unión Europea, ha cobrado especial relevancia el control y monitorización de servicios vinculados a la producción.
En concreto el punto 6 del anexo, clasifica el mayor riesgo en aquellos casos en los que éstos entran en contacto directo con el producto o con materiales y superficies que en última instancia impactan directamente en el producto.
Las instalaciones de agua, gases, vapor y aire comprimido deben por tanto diseñarse, cualificarse, mantenerse y monitorizarse adecuadamente para evitar ser causa de contaminación del producto estéril, bien sea por una pureza inadecuada, por presencia de contaminantes microbiológicos viables o por el producto de su metabolismo como pueden ser la presencia endotoxinas y pirógenos.
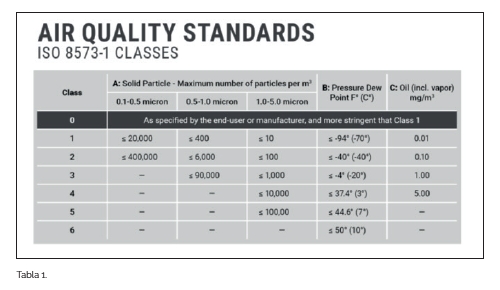
En concreto y para el aire comprimido, el marco de referencia para la pureza y su determinación lo establece la Norma ISO 8573-1:2010, y los parámetros para analizar que nos permitirán clasificar esta pureza son:
- Concentración de partículas sólidas
- Humedad residual
- Contenido en aceite
- Microorganismos viables*
*En la clasificación ISO no se incluye una numeración en cuanto a la pureza microbiológica, sin embargo, su control debe formar parte de la monitorización tal y como veremos más adelante, y es la parte 7 de la norma ISO 8573 la que recoge información específica sobre detección de agentes microbiológicos.
La calidad del aire comprimido se indica según clase numérica para cada apartado específico:
ISO 8573-1:2010 [A: B: C] donde,
- A: Partículas
- B: Humedad y agua líquida
- C: Presencia de aceite
En la producción farmacéutica, el objetivo de clase debería estar habitualmente entre 0 y 1.
Diseño de instalaciones
Una instalación en la cual durante el proceso de producción farmacéutico sea necesario que el aire/gas entre en contacto directo con el producto o bien sea utilizado en el proceso de preparación de la instalación o limpieza (esterilización, secado, fase de vaciado…), precisará de un diseño muy detallado, tanto de los circuitos de tuberías como de la calidad de estas, componentes, uniones y de los equipos de producción.
Los materiales y equipos deberán ser de la calidad y especificaciones técnicas predefinidas en la fase de diseño y deberán ser revisados previamente a su instalación, así como a posteriori en las etapas de cualificación de la instalación, operacionales y del desempeño o funcionales (IQ, OQ y PQ).
El diseño de la instalación contempla, en primer lugar, un análisis exhaustivo de las necesidades de aire comprimido de los procesos relacionados con la producción, a fin de poder definir la capacidad de producción de los equipos compresores. Definida la necesidad se deberá identificar la criticidad del aire a suministrar, decantándose o bien por la producción con un solo equipo o con equipos de back up (50/50, 70/30, 100/100).
En segundo lugar, debemos planificar adecuadamente la ubicación de la instalación, debido a que un entorno donde el aire exterior pueda estar contaminado con otros gases como hidrocarburos procedentes de la combustión de vehículos u otra maquinaria, nos hará partir de un aire de peor calidad y que no llegue a alcanzar la pureza deseada en los puntos terminales por más que nuestra instalación sea la más cara y eficiente del mercado.
En cuanto a normativas para tener en cuenta para el adecuado diseño de los componentes, contaremos con la ISO 12500 sobre filtración de aire comprimido y separadores de agua, así como con la ISO 7183 sobre el correcto secado del aire comprimido.
Por último y no menos importante, hemos de asegurarnos que durante la ejecución de la instalación se alcancen los máximos estándares, por lo que debe ser realizada por profesionales que dispongan de la formación y experiencia necesaria. Durante este proceso se deberán realizar verificaciones importantes en equipos, materiales y componentes. Algunas de las verificaciones que recomendamos y deberían ser documentadas son:
- Certificados de calidad y trazabilidad del material: acero inoxidable A316L (Norma ASME-BPE)
- Acabado superficial interno (Ra)
- Isométricos
- Certificados de los soldadores
- Estadillos diarios
- Control de soldaduras (Radiografías, boroscopiado, inspección visual…)
Los equipos deberán ser por lo tanto de alta eficiencia y en el caso que nos ocupa a nivel de la industria farmacéutica, preferiblemente compresores sin aceite, con sistemas que eviten la contaminación exterior y con sistemas de secado y filtrado que permitan alcanzar el nivel de calidad exigido en cada punto de suministro. Se deberán instalar sistemas de monitorización en línea para poder determinar y detectar en cada momento el grado de pureza de producción del aire comprimido.
Control de calidad y monitorización periódica del aire comprimido y otros gases
Para poder realizar un adecuado control de calidad hemos de tener claro qué estamos buscando y con qué herramientas contamos. El acercamiento más recomendable para ello es rodearse de un equipo multidisciplinar con conocimientos en todas las materias:
- Personal disponible con formación y experiencia en microbiología y procesos de producción específicos
- Equipo técnico a nivel de ingeniería y mantenimiento con conocimientos específicos en el ámbito de producción y tecnologías aplicables
- Estrategia de control de la contaminación (CCS)
- URS’s (Requerimientos de usuario): tanto para los límites microbiológicos como para la pureza del aire comprimido e instalaciones de producción
En cuanto a las auditorías de calidad, podemos realizar una valoración a nivel de la instalación de producción del aire comprimido o de otros gases, pero como hemos introducido previamente, la pureza del aire o gas y su calidad microbiológica son 2 conceptos diferenciados, y ésta última es probablemente la más importante en términos de seguridad en los puntos de uso directo del aire/gas en el entorno de producción o laboratorios. Por esta razón, es habitual utilizar filtros ‘microbiológicos’ de 0,22 µm en los puntos terminales y posteriormente a este filtro utilizar elementos desechables y/o esterilizables.
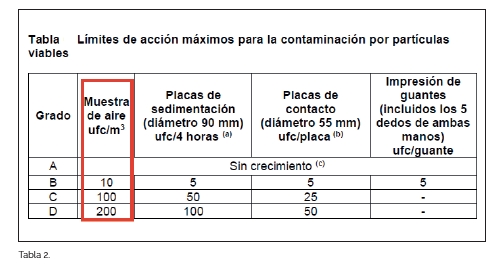
Según establece la norma, la monitorización microbiológica de salas limpias debe corresponderse con el grado de esta, por lo que según la tabla 6 de la parte 9, correspondería realizar la monitorización para un volumen concreto de aire o gas (1 m3) haciéndolo impactar sobre un medio de crecimiento específico para bacterias y hongos en placas de Petri. Los límites de detección son los siguientes, en Unidades Formadoras de Colonias (UFC):
En el entorno de producción farmacéutica es de uso habitual el aire comprimido en contacto directo con el producto además de gases como el nitrógeno, oxígeno, dióxido de carbono o argón entre otros, y tanto la pureza como la calidad microbiológica pueden ser monitorizadas en los puntos de uso.
Para esto último se ha de contar, eso sí, con la tecnología adecuada y el personal especializado en su utilización, ya que por ejemplo la conducción de un gas para su valoración microbiológica implica conocimientos sobre el propio gas y la seguridad, correcta despresurización hacia las placas de Petri donde han de impactar las partículas para ser luego cultivadas, además de en muchos casos la posterior extracción del gas o aire comprimido en condiciones de seguridad que puede incluir la adecuada filtración de alta eficacia (HEPA) u otra metodología más compleja.
Existen en el mercado tecnologías aplicables a las auditorías de calidad de pureza y de microbiología en instalaciones y en los puntos de uso, pero hemos de tener en cuenta que la reglamentación avanza y se hace cada vez más exigente. Uno de los puntos que debemos tener presentes y no perder de vista, es la obtención de datos según las normas de correcta fabricación debido a que estamos valorando elementos que forman parte del proceso de producción y pueden hacernos detener la producción con lo que ello conlleva. Aspectos como la CFR 21 parte 11 para obtención de registros electrónicos cuando sea de aplicación o la formación periódica en normas de correcta fabricación deben incluirse en las tomas de decisiones y en las evaluaciones de riesgos previas a todos los procesos de cualificación, validación y verificación o monitorizaciones periódicas.
Conclusiones
Los procesos de producción farmacéuticos son sometidos cada vez a mayores controles según avanza la tecnología en la industria.
Las instalaciones de servicios deben ser diseñadas no solamente para proporcionar el propio servicio con la calidad requerida, sino que hemos de prever el decomisionamiento tras la vida útil o las mejoras continuas objeto de los cambios regulatorios en la medida de lo posible.
Es de relevante importancia, por lo tanto, buscar el asesoramiento adecuado en las ingenierías y servicios de mantenimiento de confianza o las presentes en las propias industrias, tanto para definir la propia instalación y mantenimiento en su conjunto, así como para ayudarnos a definir la monitorización paramétrica o incluso para proporcionar este servicio si así lo incluyen.
Descarga sugerida:
Artículo escrito por:
,Linnic