La validación farmacéutica ha experimentado un cambio profundo en las últimas décadas, abandonando el enfoque tradicional en el que se trataba como una actividad final y meramente documental, para convertirse en una disciplina científica que atraviesa todas las fases del ciclo de vida del proyecto. La práctica histórica, en la que los equipos de ingeniería instalaban y ponían en marcha un sistema para que posteriormente el área de calidad iniciara su validación, generaba duplicidades, desajustes entre requisitos y entregables y retrasos significativos en el inicio de la producción.
Hoy se reconoce que estos problemas no se debían a la falta de ensayos técnicos, sino a la desconexión entre ingeniería, construcción y calidad, y especialmente a la generación tardía de evidencia regulatoria.
En la validación de hoy en día, la documentación ya no es un producto al final del proyecto, sino una construcción progresiva que refleja el pensamiento científico y la trazabilidad de decisiones, actividades y verificaciones. La evidencia se genera desde la concepción del diseño hasta la operación inicial del sistema, asegurando continuidad y consistencia entre lo que se pide, lo que se fabrica, lo que se instala y lo que se demuestra. Esto exige una trazabilidad rigurosa desde los requisitos del usuario, pasando por las decisiones de diseño y las pruebas realizadas, hasta los registros que confirman su cumplimiento. La integridad de los datos se convierte en un requisito innegociable: cada registro debe ser atribuible, fechado, verificable y reflejar las condiciones reales de la prueba.
Este enfoque implica que el área de Calidad deje de participar únicamente al final y se incorpore desde las primeras fases del proyecto. La intervención temprana de QA en la definición de requisitos, en la revisión de criterios de aceptación y en la evaluación de las estrategias de prueba permite que la evidencia generada durante todo el proyecto sea compatible con los estándares regulatorios. Este cambio, no solo reduce la necesidad de repetir pruebas, sino que también disminuye desviaciones evitables, fortalece la defensa técnica ante auditorías y acorta considerablemente los tiempos hasta la puesta en marcha.
Commissioning y el modelo integrado CQV como motor de eficiencia, evidencia representativa y solidez regulatoria
El auge del enfoque Commissioning, Qualification & Validation (CQV) ha transformado profundamente la forma de concebir la validación. No se trata de una simple reorganización de tareas, sino de una integración conceptual que reconoce que la mayor parte de la evidencia relevante para la validación se genera durante las propias actividades técnicas del proyecto. Bajo esta perspectiva, el commissioning —que comprende desde fases tempranas del proyecto hasta el ajuste funcional y operativo en condiciones reales— deja de ser una etapa puramente técnica para convertirse en el núcleo de la generación de evidencia regulatoria.
Durante las actividades de commissioning previas a la cualificación se llevan a cabo verificaciones técnicas que permiten confirmar la instalación, la integración funcional y el comportamiento general de los sistemas en su contexto operativo. Estas verificaciones proporcionan una visión temprana y fiable del desempeño real del equipo, ya que permiten observar cómo interactúan sus componentes, cómo responden los elementos de control y cómo se comportan bajo condiciones representativas.
La evidencia generada en estas etapas suele incluir registros técnicos, datos automatizados y documentación capturada en tiempo real, lo que aporta un nivel de fidelidad y coherencia particularmente valioso desde el punto de vista regulatorio. Siempre que esta evidencia cumpla criterios de integridad, trazabilidad y calidad técnica, puede constituir una base sólida para la posterior cualificación, evitando duplicidades y fortaleciendo la consistencia del expediente.
El modelo CQV sostiene que la cualificación no debe duplicar aquello que ya ha quedado verificado durante el commissioning, siempre que la evidencia cumpla los requisitos necesarios. Este cambio conceptual permite que muchas verificaciones críticas se realicen una sola vez: durante el commissioning, cuando los sistemas están vivos, ajustándose y mostrando su comportamiento real. En lugar de repetir pruebas idénticas en IQ/OQ, la cualificación se concentra únicamente en los aspectos que no pudieron evaluarse antes o que requieren condiciones operativas específicas. Esto reduce el trabajo redundante, evita retrabajos innecesarios, disminuye la probabilidad de errores derivados de reejecuciones y acorta el camino crítico del proyecto.
La integración entre commissioning y cualificación exige que ingeniería, calidad y operaciones trabajen de manera conjuntamente planificada. Antes de iniciar pruebas, los equipos acuerdan qué requisitos se verificarán en fábrica, cuáles en sitio, cuáles pueden cubrirse durante commissioning y cuáles requieren pruebas específicas en las fases finales. Este acuerdo temprano elimina ambigüedades, define responsabilidades, evita solapamientos y asegura que cada prueba tenga un propósito científico claro. El resultado es un proceso de validación donde la evidencia se genera cuando aporta más valor, con un nivel de detalle superior y con mayor capacidad para demostrar el cumplimiento regulatorio.
Validación basada en riesgo en la era del CQV: eficiencia, resiliencia y expedientes regulatorios más sólidos
La validación basada en riesgo constituye hoy el núcleo del pensamiento regulatorio moderno. Este enfoque reconoce que no todos los sistemas ni todas las funciones impactan de la misma manera en la calidad del producto, y que las actividades de verificación deben priorizarse en función de su criticidad. La clasificación entre sistemas de impacto directo e impacto indirecto permite asignar recursos, y darles profundidad documental y esfuerzo de verificación de forma proporcional al riesgo real asociado a cada elemento. Un sistema de impacto directo es aquel que afecta directamente a la calidad del producto final; mientras que un sistema de impacto indirecto influye en el entorno o en la operación, pero sin un vínculo directo con el producto final. Esta distinción orienta de manera inmediata qué aspectos requieren mayor rigor documental, qué pruebas deben ser formales y dónde se puede aplicar una verificación técnica más simplificada.
La integración del enfoque CQV con la validación basada en riesgo amplifica el valor de este modelo. Cuando se identifican los requisitos críticos —especialmente aquellos de impacto directo—, el enfoque CQV permite verificar muchos de ellos en etapas tempranas mediante actividades técnicas ya previstas en el ciclo del proyecto. Esto elimina el riesgo de que los elementos críticos permanezcan sin verificar hasta fases tardías, mejora la capacidad de detección de desviaciones antes de que se conviertan en problemas mayores y reduce la probabilidad de que aparezcan barreras temporales en la fase de cualificación. Por su parte, los sistemas de impacto indirecto pueden gestionarse de forma más eficiente, con verificaciones proporcionadas a su relevancia y sin necesidad de formalismos innecesarios.
La convergencia entre CQV y la validación basada en riesgo produce un impacto significativo en la eficiencia global del proyecto. La evidencia se genera cuando aporta más información y bajo condiciones que reflejan fielmente el comportamiento real de los sistemas; la documentación se estructura de forma lógica y coherente; las desviaciones se detectan en momentos en los que su resolución es más rápida y menos disruptiva, y la fase de cualificación se vuelve más ágil al centrarse exclusivamente en los elementos que no pueden verificarse antes. Esta integración contribuye a proyectos con mayor predictibilidad, expedientes más defendibles y un uso más inteligente de los recursos, todo ello sin comprometer —y a menudo reforzando— la seguridad del paciente y la integridad del producto. En conjunto, la validación basada en riesgo y el enfoque integrado CQV constituyen un marco técnico en el que ciencia, ingeniería y calidad trabajan de forma coordinada para generar evidencia sólida, trazable y eficiente, garantizando sistemas robustos y preparados frente a auditorías regulatorias sin caer en prácticas redundantes o excesivamente administrativas.
Fases del proceso
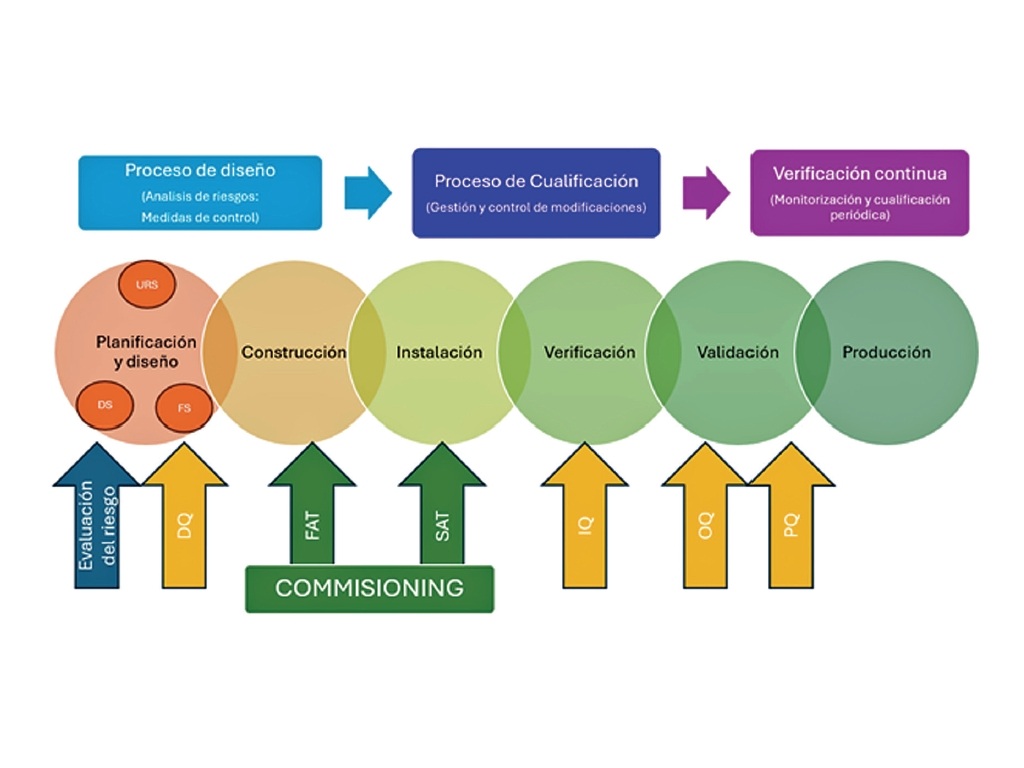
La primera fase, Planificación Estratégica del Programa CQV, establece los cimientos del proyecto mediante una evaluación global de instalaciones, utilidades, equipos y procesos, acompañada de un análisis de riesgos que identifica los sistemas críticos. A partir de ello y se definen los User Requirements Specifications (URS) y se elabora el Validation Master Plan (VMP), que guían todo el diseño posterior.
En la segunda etapa, el Commissioning, se verifican técnicamente los sistemas desde su diseño hasta su puesta en marcha inicial. Las revisiones de diseño, las pruebas FAT y SAT y las verificaciones funcionales tempranas permiten confirmar que la instalación cumple los requisitos previstos y está lista para su cualificación formal.
La tercera fase, la Cualificación, constituye el núcleo del cumplimiento regulatorio. A través de DQ, IQ, OQ y PQ se demuestra que el diseño es adecuado, la instalación correcta, la operación conforme a los rangos esperados y el desempeño consistente bajo condiciones reales. El informe de cualificación consolida los resultados y habilita el sistema para su uso regulado.
A continuación, la fase de Validación aborda la verificación del proceso en su conjunto, incluyendo validación de procesos, limpieza y sistemas informatizados, junto con la verificación continua que garantiza la robustez a lo largo del tiempo.
El Mantenimiento del Estado Validado asegura la sostenibilidad del sistema mediante control de cambios, re-cualificaciones, auditorías y una gestión rigurosa de la documentación.
Finalmente, la etapa de Cierre e Inicio de la Operación Estándar formaliza la transición a la operación rutinaria, integrando mantenimiento, calibración, gestión de desviaciones y la formación del personal, consolidando un estado validado plenamente operativo.
Descarga sugerida:
Artículo escrito por:
Ainhoa García Cassinello y Paulino Pastor
Commissioning Manager del área farmacéutica y Director general, respectivamente
Commtech Commissioning Services (compañía del Grupo Aire Limpio) y Ambisalud GMP Consulting (compañía del Grupo Aire Limpio), respectivamente