Dentro de la industria farmacéutica, si hablamos de buenas prácticas de manufactura (BPM) y de su cumplimiento, es porque está implícito un sistema de gestión de calidad (SGC) debidamente establecido, implementado y que demuestra día a día su robustez, adecuación, conveniencia y eficacia.
No se puede hablar de calidad, seguridad y eficacia si, tanto en las BPM como en el SGC involucrado, no se demuestra cumplimiento con evidencia objetiva y dentro de ellos hay 3 sistemas que son fundamentales para lograr los objetivos técnicos y regulatorios. Nos referimos al sistema de gestión del riesgo (SGR), control de cambios (CC) y no conformidades (NC) o desviaciones.
En este artículo se manejarán las siguientes definiciones tomadas de diferentes regulaciones y guías farmacéuticas:
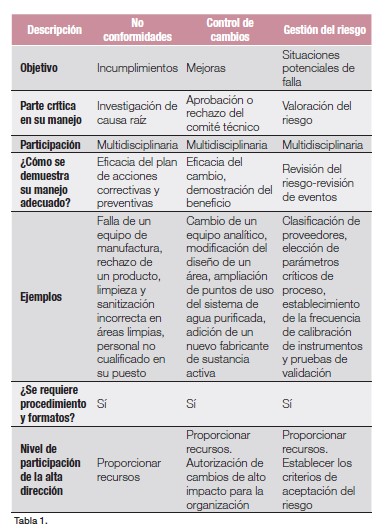
• No conformidad o desviación. Incumplimiento de un requisito. No cumplimiento de un requisito previamente establecido. No satisfacción de un requisito. Deficiencia en una característica, especificación de un producto, parámetro de proceso, registro o procedimiento que hace que la calidad de un producto sea inaceptable, indeterminada o que no está de acuerdo con requerimientos especificados.
• Control de cambios. Evaluación y documentación de cualquier cambio que puede impactar en la calidad del producto. Conjunto de actividades documentadas que especifican los pasos para incorporar modificaciones previstas y que pueden impactar la calidad y desempeño del producto. Sistema formal, por el cual, representantes calificados de las disciplinas apropiadas evalúan los cambios propuestos o realizados que pudieran afectar el estado validado/calificado, la estabilidad, la intercambiabilidad o alguna condición de autorización sanitaria.
• Gestión del riesgo. Aplicación sistemática de políticas, procedimientos y prácticas de gestión de calidad para la valoración, control, comunicación y revisión de riesgos. Enfoque sistemático para la identificación, análisis, evaluación, control, comunicación y revisión de los riesgos en los procesos y productos o servicios a través de su ciclo de vida. Actividades coordinadas para dirigir y controlar a una organización con respecto al riesgo.
Sin duda alguna, hay otros elementos del SGC que también son importantes. Sin embargo, en este artículo nos centraremos en estos 3 en particular por la confusión constante en su aplicación ya que, al estar relacionados, puede ser fácil irse por la puerta equivocada y pensar que se pueden utilizar indistintamente.
Tanto gestión de riesgos, como no conformidades y control de cambios son exigidos en la actualidad y no solo se trata de tener procedimientos documentados, sino de demostrar su eficacia y que el entendimiento está presente entre todo el personal.
La unidad de calidad generalmente es la que gestiona su cumplimiento, aunque esto no significa que sea la única responsable de ejecutarlos y documentarlos.
Bajo un contexto histórico, la gestión del riesgo es el último sistema en exigirse a nivel regulatorio, en comparación con no conformidades y control de cambios, el cual tomó más fuerza junto con la implementación de los sistemas de validación.
Esto lleva a reflexionar también sobre la coherencia de los requisitos regulatorios, en los cuales, para poder ser exitoso, se debe contar con un sistema de gestión de calidad con los elementos básicos presentes, para que este último requisito de gestión del riesgo sea un éxito.
¿Cómo lo veo?
El manejo adecuado en los sistemas previos de no conformidades y control de cambios, reflejan la madurez y cultura que se tendrá en el sistema de gestión de riesgos. Las debilidades, deficiencias, olvidos, supuestos y demás fallas que nos llevan al fracaso en NC y CC, también se reflejaran en el SGR.
La cuestión es cómo exigir a una organización que se preocupe por algo que no ha pasado (enfoque de riesgo) si está lleno de ‘problemas’, entre los que se encuentran, por ejemplo, procesos y trabajos repetidos, recuperaciones, producto no conforme, rechazos, resultados fuera de especificaciones, quejas, devoluciones, retiros de producto, hallazgos negativos en una auditoria o deficiencias en una inspección sanitaria, entre otros.
Recordemos una definición de riesgo con enfoque hacia las BPM: combinación de la probabilidad de que se presente un daño y el impacto de este daño.
Como se puede observar claramente, para gestionar el riesgo debemos centrarnos en la probabilidad de un daño (daño al paciente, pero también las empresas consideran daño a los procesos, sistemas, equipos, instalaciones, servicios o materiales, por ejemplo), la consecuencia (también denominada severidad, impacto, gravedad, efecto) y, altamente recomendable, la capacidad de detección (dependiendo de la herramienta de gestión de riesgo que se utilice).
Si se ponen los recursos en una balanza, estos serán destinados por lógica y prioridad a situaciones que ya hayan pasado y, sobre todo, que requieran atenderse por la necesidad de liberar un producto al mercado (por ejemplo, una no conformidad que requiera respuesta inmediata) o de dar una respuesta a la agencia sanitaria como resultado de una inspección.
Esto ocasiona que, tal vez sin darnos cuenta, se vaya dejando de lado el objetivo y la aplicación del sistema de control de cambios y gestión de riesgo. También que sean solo procedimientos que se muestran en una auditoria o inspección, pero que en la práctica no se utilizan, salvo cuando el SGC lo exija con precisión. Es el caso de, por ejemplo, aplicar la gestión de riesgo para selección de las pruebas a colocar en un protocolo de calificación de un equipo de manufactura.
Las claves para poder diferenciar las aplicaciones de los 3 sistemas e identificar sus similitudes están en la tabla 1.
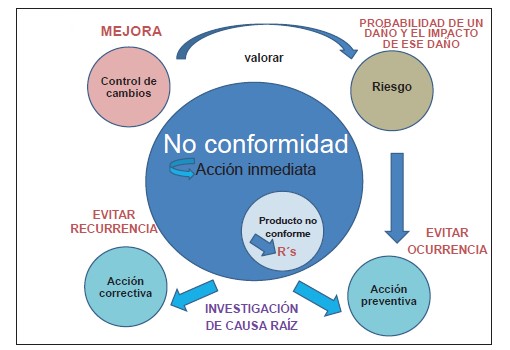
Asimismo, hay interrelación entre ellos, lo cual puede explicarse con la figura 1.
No conformidad
• Dentro del concepto de no conformidad, se encuentra incluido el producto no conforme. Pudiera explicarse indicando que: todo producto no conforme es una no conformidad, pero no toda no conformidad es un producto no conforme.
• Recordar que no todo producto no conforme es automáticamente un rechazo, ya que (previo análisis del comité evaluador) es posible encontrar una solución, considerando la complejidad, naturaleza del producto, etapa, forma farmacéutica y requisitos regulatorios. Es lo que se conoce como las R´s: reproceso, retrabajo, recuperación, reacondicionamiento y rechazo.
• Es ampliamente útil clasificar las no conformidades con un enfoque de riesgo. Esto no significa que se confunda riesgo con no conformidad. Quiere decir que las no conformidades deberían tomar en cuenta, para su clasificación, no solo su impacto o su gravedad, sino también las veces que se ha presentado en cierto periodo de tiempo indicado.
• Considerando su clasificación, se debería dejar estipulado el alcance del programa de acciones inmediatas (también de denominadas correcciones o acciones remediadoras), acciones correctivas y acciones preventivas (también conocido como CAPA por sus siglas en inglés), así como la correspondiente investigación de la causa raíz.
Todo esto, de acuerdo con las siguientes definiciones:
• Corrección (acción inmediata): acción para eliminar una no conformidad detectada. Puede realizarse con anterioridad, simultáneamente o después de una acción correctiva. Reparación o ajuste relacionado para la disposición de una no conformidad existente. Algunos la llaman acción de contención.
• Acción correctiva: acción realizada para eliminar la causa de una no conformidad (ya sucedió) y evitar que vuelva a ocurrir (recurrencia).
• Acción preventiva: acción realizada para eliminar la causa de una no conformidad potencial u otra situación potencial no deseada. Es decir, algo que no ha sucedido, pero puede suceder una vez que se analiza la no conformidad que ya se presentó (por ejemplo, se tuvo un incumplimiento en el equipo 1 y es factible que llegará a suceder en el equipo 2) y evitar que ocurra. En este caso, aunque su enfoque es hacia una situación potencial y pudiera tener un enfoque de riesgo, esta acción se presentó o fue establecida hasta que se tuvo como antecedente una no conformidad.
Control de cambios
• Los cambios principalmente se enfocan en mejoras por calidad y/o productividad.
• Si bien hay que hacer cambios por actualizaciones en las regulaciones, se entiende que las agencias sanitarias hacen esos cambios por mejorar el cumplimiento de las BPM y disminuir los riesgos implícitos.
• Cambios no planeados se consideran no conformidades.
• Las propuestas de cambio pueden ser aprobadas o rechazadas por un comité técnico de acuerdo con la justificación de la propuesta, que necesita ser valorada por los riesgos que puede traer, considerando que al momento de presentar la solicitud de cambio, no se está en incumplimiento. Esto significa que se ha alcanzado un estado de control que debería cuidarse y que cualquier modificación requiere una evaluación detallada de su impacto en diferentes escenarios, donde la valoración del riesgo es fundamental para la toma adecuada de decisiones.
• Algunas acciones correctivas y preventivas derivadas de no conformidades pueden incluir en su naturaleza cambios, como un cambio de equipo o de proveedor de materia prima, si en la investigación inicial salen como causa de la desviación y, por tanto, requieren modificarse. Sin embargo, este cambio surgió como parte de un incumplimiento, no como una propuesta de mejora, y deberían estar documentados en el programa CAPA. Hay organizaciones que reconocen que habrá cambios derivados de CAPA cuya complejidad, lógica y secuencia no puede ser manejada tan a detalle en los formatos de CAPA, que tienden a ser más simples y concretos que los formatos de control de cambios y documentan estos CAPA en un formato de control de cambios. Esto es aceptable si en ningún momento se elimina el registro como CAPA y se reconoce que el formato de control de cambios se utiliza solo para un mejor manejo de las acciones y no se desconoce que proviene de una no conformidad. Tanto el programa CAPA como el control de cambios tendrán que ser cerrados documentalmente en paralelo.
Gestión del riesgo
• Su aplicación principal es con un enfoque proactivo.
• No se debe utilizar la gestión de riesgo para ocultar noconformidades, evitar su documentación, pretender justificar no cumplir los requisitos regulatorios o sustituir el registro de los controles de cambios o programas CAPA.
• Se tienen diferentes herramientas para poder llevar a cabo la gestión del riesgo y que permiten documentar eficazmente todas las etapas.
• La gestión del riesgo parte de una situación potencial, no de un incumplimiento. Por tanto, las acciones que en algún momento se llegaran a requerir para controlar el riesgo tienen una naturaleza preventiva. Pero, a diferencia de la acción preventiva derivada de la no conformidad, estas acciones de control de riesgo no tienen este origen y representan el enfoque actual de las agencias sanitarias: el preocuparse por lo que no ha sucedido y actuar en consecuencia y no esperar a tener algún incumplimiento.
Por lo tanto, podemos concluir que los 3 sistemas son críticos y fundamentales dentro de un sistema de gestión de calidad y no se debe confundir en ningún momento su objetivo y aplicación. Un buen manejo de un sistema de no conformidades y CAPA traerá a largo plazo la disminución de los incumplimientos, dejando el principal uso de los recursos en el enfoque de mejoras (control de cambios) y gestiones del riesgo. Mientras tanto, dependiendo de la madurez del sistema de gestión de calidad, habrá que mantener la balanza con los enfoques adecuados.
Artículo escrito por:
Elizabeth Martínez Flores
Gmp/Gxp Coach, Consultora e Instructora Internacional. Directora General
,Grupo Terra Farma